> جين كيربي
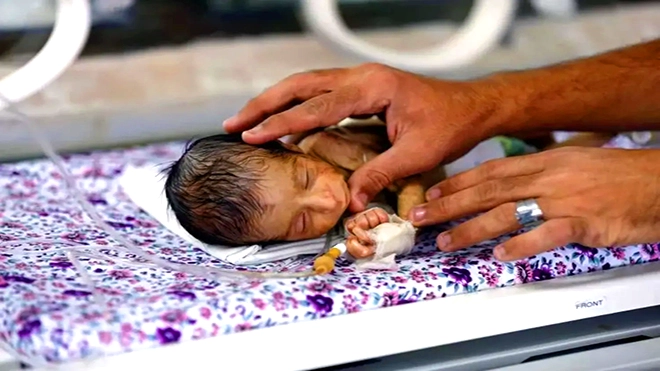
أعطت هيئة تنظيم استخدام الأدوية في المملكة المتحدة ترخيصًا لأول علاج جيني في العالم بوصفه علاجًا محتملًا لاثنين من اضطرابات الدم الموروثة.
ويعد دواء "كاسجيفي" Casgevy المخصص لمرضى فقر الدم المنجلي وثلاسيميا من نوع بيتا، أول علاج جيني ينال ترخيصًا، مع الإشارة إلى أنه يحتوي على أداة تعديل الجينات المعروفة باسم "كريسبر" Crispr، التي حاز مخترعوها على جائزة نوبل في عام 2020.
وفي تفاصيل متصلة، أوضحت "وكالة تنظيم استخدام الأدوية ومنتجات الرعاية الصحية" Medicines and Healthcare Products Regulatory Agency في بريطانيا، أن ذلك العلاج الجيني خصص للمرضى الذين تبلغ أعمارهم 12 سنة أو أكثر، "عقب تقييم دقيق لسلامته وجودته وفعاليته".
وينظر إلى مرضي فقر الدم المنجلي وثلاسيميا بيتا، باعتبارهما حالتين وراثيتين ناجمتين عن خلل جيني في تركيب بروتين "هيموجلوبين" الذي تستخدمه خلايا الدم الحمراء في نقل الأوكسجين إلى جميع أنحاء الجسم.
وكذلك يعتبر فقر الدم المنجلي مرضًا شائعًا بشكل خاص في صفوف من جذور عائلية ترجع إلى أفريقيا أو منطقة البحر الكاريبي. وبشكل رئيس، يؤثر مرض ثلاسيميا من النوع بيتا، على أناس كثيرين في منطقة البحر الأبيض المتوسط، وجنوب آسيا، وجنوب شرقي آسيا، والشرق الأوسط.
وقد تشمل أعراض مرض فقر الدم المنجلي معاناة آلام شديدة جدًا والتهابات خطرة تهدد الحياة، إضافة إلى فقر الدم [أي حدوث خفض في مستوى الحديد في الدم، مع ملاحظة أنه مكون أساس للهيموجلوبين]. ويعاني الأشخاص المصابون بمرض ثلاسيميا بيتا فقر الدم الشديد. وفي الغالب، يحتاج المصابون بذلك المرض إلى نقل دم كل ثلاثة إلى خمسة أسابيع، إلى جانب أدوية وحقن تعطى بشكل منتظم.
واستطرادًا، جرى تصميم دواء "كاسجيفي" كي يعمل من طريق تعديل الخلل في تركيب جين معين يوجد في الخلايا الجذعية التي تنتج في نخاع العظم، مما يؤدي إلى إنتاج هيموجلوبين صحي وفاعل.
وفي سياق تلك العملية، يعمد الأطباء إلى استخراج الخلايا الجذعية من نخاع العظم، ثم تعديلها في المختبر، وبعدها يعاد غرس الخلايا المصححة في النخاع العظمي للمريض، ما يساعد في شفاء مصابين بالثلاسيميا من النوع بيتا.
حتى الآن، تمثل الخيار العلاجي الدائم الوحيد بنقل نخاع العظم ممن يتوافق ذلك النسيج لديهم مع نظيره لدى المريض، على رغم تضمنه إمكانية تعرضه للرفض من جسم المريض جوليان بيتش.
وفي سياق متصل، أوضحت "وكالة تنظيم استخدام الأدوية ومنتجات الرعاية الصحية" أن التجارب السريرية وجدت أن "كاسجيفي" أعاد إنتاج الهيموجلوبين الصحي لدى غالبية من طبق عليهم ذلك العلاج.
وتوضيحًا، أظهرت التجارب السريرية على مرض فقر الدم المنجلي، أن 28 من أصل 29 مريضًا في تحليل شمل 97 % من العينة، لم يشعروا بآلام شديدة طوال 12 شهرًا في الأقل بعد العلاج.
وفي المقابل، بينت التجارب السريرية لثلاسيميا بيتا، أن 39 من أصل 42 مريضًا في تحليل شمل 93 % منهم، لم يكونوا في حاجة إلى نقل خلايا الدم الحمراء طوال 12 شهرًا في الأقل بعد العلاج. وقد حدث خفض في الحاجة إلى عمليات نقل الخلايا الحمراء بقرابة 70 %، لدى 3 % المتبقية من أولئك المرضى.
وفي تعليق بارز، أوضح المدير التنفيذي الموقت لجودة الرعاية الصحية والوصول إليها لدى "وكالة تنظيم استخدام الأدوية ومنتجات الرعاية الصحية"، جوليان بيتش، إن "فقر الدم المنجلي وثلاسيميا بيتا مرضان مؤلمان يستمران مدى الحياة، ويمكن أن يتسببا بالوفاة في بعض الحالات". وأضاف "حتى الآن، تمثل الخيار العلاجي الدائم الوحيد بنقل نخاع العظم ممن يتوافق ذلك النسيج لديهم مع نظيره لدى المريض، على رغم تضمنه إمكانية تعرضه للرفض من جسم المريض".
ثمة مجموعة محدودة من الأدوية متاحة للمرضى في الحاضر، لذلك أرحب بأخبار أتت اليوم وخلاصتها الحكم على علاج جديد بأنه آمن وفعال، ولديه القدرة على تحسين نوعية حياة أشخاص كثيرين وبشكل كبير.
جون جيمس
وكذلك ذكر جون جيمس، الرئيس التنفيذي لجمعية فقر الدم المنجلي، أن "اضطراب فقر الدم المنجلي يشكل حالة منهكة للغاية، ويسبب مجموعة من الآلام الشديدة لدى الأشخاص الذين يصيبهم، مع إمكانية أن يؤدي إلى الوفاة المبكرة". وأضاف "ثمة مجموعة محدودة من الأدوية متاحة للمرضى في الحاضر، لذلك أرحب بأخبار أتت اليوم وخلاصتها الحكم على علاج جديد بأنه آمن وفعال، ولديه القدرة على تحسين نوعية حياة أشخاص كثيرين وبشكل كبير".
وحتى الآن، لم يجر اختبار فاعلية علاج "كاسجيفي" للاستخدام على نطاق واسع من قبل هيئة "الخدمات الصحية الوطنية".
"إندبندنت عربية".
ويعد دواء "كاسجيفي" Casgevy المخصص لمرضى فقر الدم المنجلي وثلاسيميا من نوع بيتا، أول علاج جيني ينال ترخيصًا، مع الإشارة إلى أنه يحتوي على أداة تعديل الجينات المعروفة باسم "كريسبر" Crispr، التي حاز مخترعوها على جائزة نوبل في عام 2020.
وفي تفاصيل متصلة، أوضحت "وكالة تنظيم استخدام الأدوية ومنتجات الرعاية الصحية" Medicines and Healthcare Products Regulatory Agency في بريطانيا، أن ذلك العلاج الجيني خصص للمرضى الذين تبلغ أعمارهم 12 سنة أو أكثر، "عقب تقييم دقيق لسلامته وجودته وفعاليته".
وينظر إلى مرضي فقر الدم المنجلي وثلاسيميا بيتا، باعتبارهما حالتين وراثيتين ناجمتين عن خلل جيني في تركيب بروتين "هيموجلوبين" الذي تستخدمه خلايا الدم الحمراء في نقل الأوكسجين إلى جميع أنحاء الجسم.
وكذلك يعتبر فقر الدم المنجلي مرضًا شائعًا بشكل خاص في صفوف من جذور عائلية ترجع إلى أفريقيا أو منطقة البحر الكاريبي. وبشكل رئيس، يؤثر مرض ثلاسيميا من النوع بيتا، على أناس كثيرين في منطقة البحر الأبيض المتوسط، وجنوب آسيا، وجنوب شرقي آسيا، والشرق الأوسط.
وقد تشمل أعراض مرض فقر الدم المنجلي معاناة آلام شديدة جدًا والتهابات خطرة تهدد الحياة، إضافة إلى فقر الدم [أي حدوث خفض في مستوى الحديد في الدم، مع ملاحظة أنه مكون أساس للهيموجلوبين]. ويعاني الأشخاص المصابون بمرض ثلاسيميا بيتا فقر الدم الشديد. وفي الغالب، يحتاج المصابون بذلك المرض إلى نقل دم كل ثلاثة إلى خمسة أسابيع، إلى جانب أدوية وحقن تعطى بشكل منتظم.
واستطرادًا، جرى تصميم دواء "كاسجيفي" كي يعمل من طريق تعديل الخلل في تركيب جين معين يوجد في الخلايا الجذعية التي تنتج في نخاع العظم، مما يؤدي إلى إنتاج هيموجلوبين صحي وفاعل.
وفي سياق تلك العملية، يعمد الأطباء إلى استخراج الخلايا الجذعية من نخاع العظم، ثم تعديلها في المختبر، وبعدها يعاد غرس الخلايا المصححة في النخاع العظمي للمريض، ما يساعد في شفاء مصابين بالثلاسيميا من النوع بيتا.
حتى الآن، تمثل الخيار العلاجي الدائم الوحيد بنقل نخاع العظم ممن يتوافق ذلك النسيج لديهم مع نظيره لدى المريض، على رغم تضمنه إمكانية تعرضه للرفض من جسم المريض جوليان بيتش.
وفي سياق متصل، أوضحت "وكالة تنظيم استخدام الأدوية ومنتجات الرعاية الصحية" أن التجارب السريرية وجدت أن "كاسجيفي" أعاد إنتاج الهيموجلوبين الصحي لدى غالبية من طبق عليهم ذلك العلاج.
وتوضيحًا، أظهرت التجارب السريرية على مرض فقر الدم المنجلي، أن 28 من أصل 29 مريضًا في تحليل شمل 97 % من العينة، لم يشعروا بآلام شديدة طوال 12 شهرًا في الأقل بعد العلاج.
وفي المقابل، بينت التجارب السريرية لثلاسيميا بيتا، أن 39 من أصل 42 مريضًا في تحليل شمل 93 % منهم، لم يكونوا في حاجة إلى نقل خلايا الدم الحمراء طوال 12 شهرًا في الأقل بعد العلاج. وقد حدث خفض في الحاجة إلى عمليات نقل الخلايا الحمراء بقرابة 70 %، لدى 3 % المتبقية من أولئك المرضى.
وفي تعليق بارز، أوضح المدير التنفيذي الموقت لجودة الرعاية الصحية والوصول إليها لدى "وكالة تنظيم استخدام الأدوية ومنتجات الرعاية الصحية"، جوليان بيتش، إن "فقر الدم المنجلي وثلاسيميا بيتا مرضان مؤلمان يستمران مدى الحياة، ويمكن أن يتسببا بالوفاة في بعض الحالات". وأضاف "حتى الآن، تمثل الخيار العلاجي الدائم الوحيد بنقل نخاع العظم ممن يتوافق ذلك النسيج لديهم مع نظيره لدى المريض، على رغم تضمنه إمكانية تعرضه للرفض من جسم المريض".
ثمة مجموعة محدودة من الأدوية متاحة للمرضى في الحاضر، لذلك أرحب بأخبار أتت اليوم وخلاصتها الحكم على علاج جديد بأنه آمن وفعال، ولديه القدرة على تحسين نوعية حياة أشخاص كثيرين وبشكل كبير.
جون جيمس
وكذلك ذكر جون جيمس، الرئيس التنفيذي لجمعية فقر الدم المنجلي، أن "اضطراب فقر الدم المنجلي يشكل حالة منهكة للغاية، ويسبب مجموعة من الآلام الشديدة لدى الأشخاص الذين يصيبهم، مع إمكانية أن يؤدي إلى الوفاة المبكرة". وأضاف "ثمة مجموعة محدودة من الأدوية متاحة للمرضى في الحاضر، لذلك أرحب بأخبار أتت اليوم وخلاصتها الحكم على علاج جديد بأنه آمن وفعال، ولديه القدرة على تحسين نوعية حياة أشخاص كثيرين وبشكل كبير".
وحتى الآن، لم يجر اختبار فاعلية علاج "كاسجيفي" للاستخدام على نطاق واسع من قبل هيئة "الخدمات الصحية الوطنية".
"إندبندنت عربية".